Researchers in CIPHER and MIT Lincoln Laboratory Achieve High Scalability in the Molecular Modeling of SARS-CoV-2
In a publication released this week, Dr. Colby T. Ford of the UNC Charlotte CIPHER Center and researchers from MIT Lincoln Laboratory showcased immense scalability in modeling the interaction of human cytokines against coronavirus nucleocapsid proteins using high-performance computing.
Background
Cytokines are small proteins that are responsible for modulating immune responses in the body, responding to infections, trauma, cancer, etc. They induce responses such as inflammation, which can help fight infections. However, if this response gets out of control, it can be deadly.
In COVID-19 infections, our bodies respond the SARS-CoV-2 virus through various means, some of which include cytokines. It is thought that some of these interactions of SARS-CoV-2 proteins with human immune proteins could be the cause of “long COVID” or severe reactions to the infection in general. Thus, the goal of this study was to computationally model such interactions and posit which, if any, may be important in future interventions.
Experimental Overload
In this study, the authors used 64 human cytokines and 17 coronavirus nucleocapsid (N) proteins (from coronaviruses such as MERS, SARS-CoV, and multiple SARS-CoV-2 variants) to generate 1,088 molecular modeling experiments (64 x 17). The goal was to learn if there are differences in how various cytokines interact with certain N proteins and if this interaction has gotten stronger/weaker in new SARS-CoV-2 variants as compared to other coronaviruses.
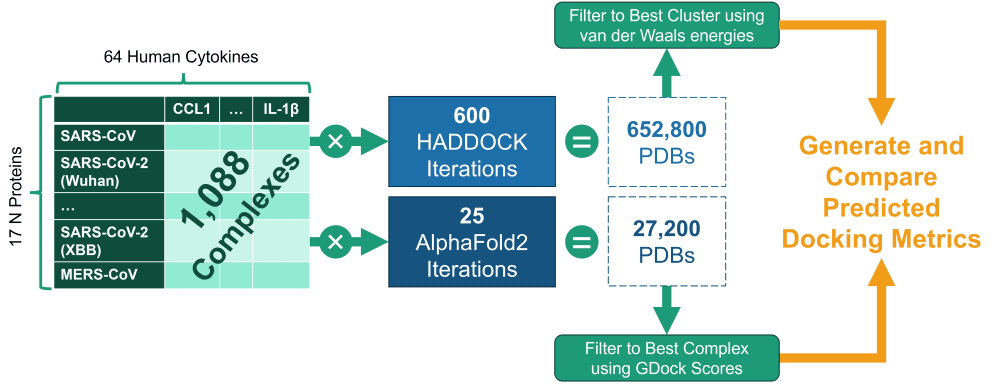
Each N <> cytokine pair was run through AlphaFold2- multimer, an AI-based took for predicting protein-protein interactions, and HADDOCK, a semi-physicochemical modeling suite.
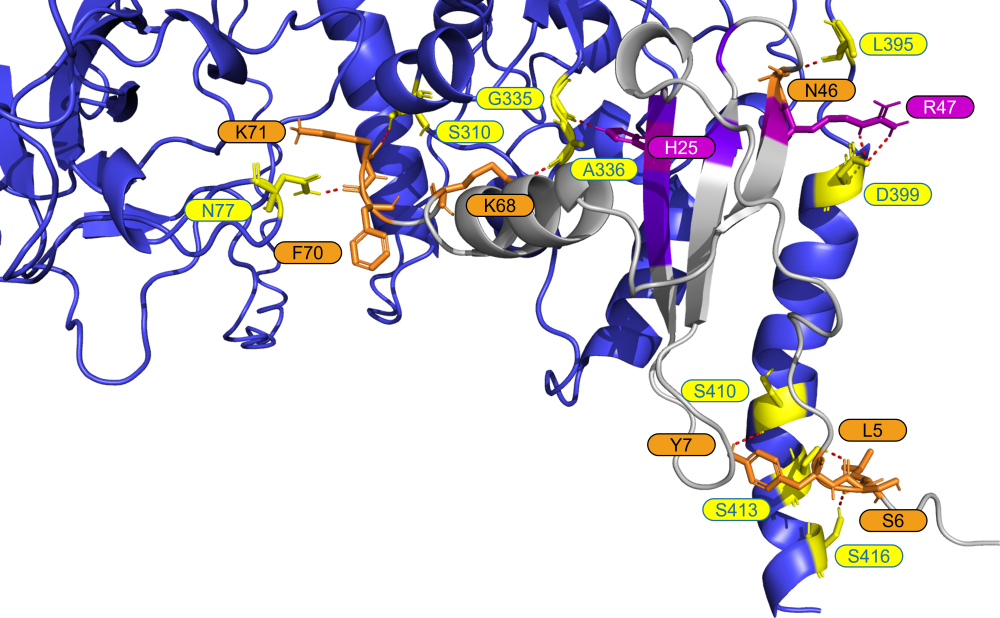
For each N <> cytokine experiment, predicted protein complexes were generated and visualized to better understand the binding location of the proteins to one another.
This process produced 100s of thousands of PDB structures, which were then evaluated and filtered programmatically using the binding affinity metrics (e.g., van der Waals energy, Gibbs energy, electrostatic energy, buried surface area), resulting in the best predicted complexes for each experiment.
What Did We Learn?
Scientifically…
This in silico study matches the results of two empirical studies (López-Muñoz et al., 2022 and López-Muñoz et al., 2023) where numerous cytokines were found to bind with high affinity in SARS-CoV-2. However, there were additional “hits” found in this study that suggest there are more cytokines being interfaced during a COVID-19 infection.
In addition, the authors showed that there are multiple cytokines for which the predicted interaction with coronavirus N proteins has weakened or strengthened over time.
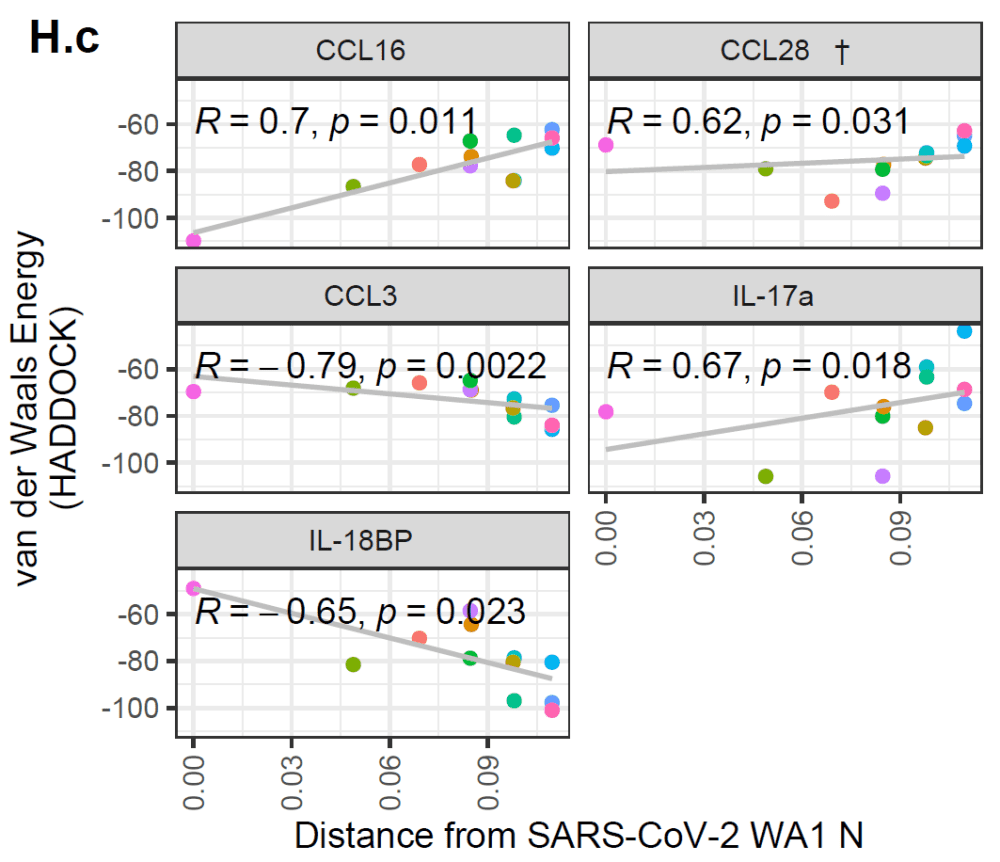
An increase in these metrics would indicate a worse binding over time (and vice versa).
“These findings offer valuable insights into the molecular mechanisms underlying viral pathogenesis and may guide the future development of targeted interventions.”
Technically…
As part of this study, the researchers ran AlphaFold2-multimer and HADDOCK, two molecular docking tools, on various types of compute nodes in a high-performance computing (HPC) cluster.
The article describes the variation in the predicted protein-protein interactions in the AlphaFold2-multimer outputs versus the HADDOCK outputs. Furthermore, performance benchmarks are shown that indicate ROI for using larger quantities of CPU and GPU cores and that core speed matters.

“The high-performance computational framework presented here will not only aid in accelerating the discovery of effective interventions against emerging viral threats, but extend to other applications of high throughput protein-protein screens.”
Learn More:
Read the article in Frontiers in Bioinformatics here: Frontiers | Human Cytokine and Coronavirus Nucleocapsid Protein Interactivity Using Large-Scale Virtual Screens (frontiersin.org)
Resources:
- GitHub Repository: https://github.com/tuplexyz/SARS-CoV-2_N-Cytokine_Docking
- HADDOCK Docker Image: https://github.com/colbyford/HADDOCKer
Disclaimer
MIT Lincoln Lab DISTRIBUTION STATEMENT A. Approved for public release.
Distribution is unlimited. This material is based upon work supported by the Department of the Air Force under Air Force Contract No. FA8702-15-D-0001. Any opinions, findings, conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the Department of the Air Force.